
Bioinformatyka wspierana matematyką

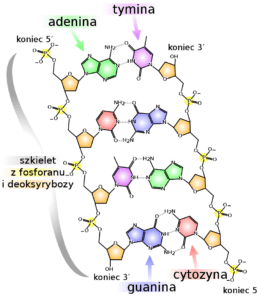
Rysunek podwójnej helisy DNA publikujemy za zgodą prof. Jacka Błażewicza1.
Bioinformatyka jest jedną z najmłodszych nauk, której burzliwy rozwój został wymuszony przez postęp w dziedzinie nauk biologicznych, a umożliwiły go dokonane przełomowe osiągnięcia matematyków zajmujących się algorytmami kombinatorycznymi i ich wdrożenia w samej informatyce.1
Wielu badaczy, mówiąc o bioinformatyce, ma na myśli głównie aspekty związane z biologią na poziomie molekularnym (DNA, RNA, białko). Stymulującym to odkryciem było podanie w 1953 r. przez Watsona i Cricka (razem ze współtwórcami Wilkinsem i Franklin nagrodzonymi Noblem w 1962 r.) modelu podwójnej helisy łańcucha DNA, przechowującego (kodującego) informację genetyczną we wszystkich organizmach żywych, co barwnie przedstawia jeden z autorów w swej opowieści.2
| Sklejanie DNA – zadanie z konkursu matematyczno-informatycznego KOALA (4. edycja)Ludzka nić DNA liczy ok. 3 mld elementów, czyli cząstek kwasu dezoksyrybonukleinowego. Cząstka może mieć jedną z czterech wartości: G, T, C lub A.
Odczytanie tak długiego łańcucha jest bardzo kłopotliwe. Ponieważ łatwo jest odczytać krótkie łańcuchy, więc jedną z metod odcyfrowania DNA jest podzielenie łańcucha, a ściślej wielu kopii tego samego łańcucha w roztworze, na mniejsze odcinki. Podziały dla poszczególnych kopii mogą być różne i np. jedna kopia łańcucha ACTACAG może zostać podzielona na odcinki: ACT, AC, AG, a inna kopia na odcinki: AC, TA, CAG. Wszystkie tak powstałe odcinki współistnieją w tym samym roztworze. Później wczytuje się informacje o odcinkach do pamięci programu komputerowego, który zajmuje się odtworzeniem prawdopodobnego wyglądu wyjściowego łańcucha. Komputer szuka takiego łańcucha, który zawiera wszystkie odcinki z roztworu, a jednocześnie jego długość jest jak najmniejsza. Na przykład najkrótszym łańcuchem zawierającym wszystkie odcinki: AC, GTA, CC, TAC jest przykładowo GTACC. Kolejność odczytu jest ważna i łańcuch CCATG nie byłby dobrym rozwiązaniem. Jaka jest najmniejsza możliwa długość łańcucha zawierającego wszystkie odcinki: ACTA,CTAT, CGAC, ATACGA, ACGA, TACG, GACTA, TATA? |
Zadanie pt. Sklejanie DNA, zapisane powyżej, to przykład problemu biologicznego polegającego na odczytaniu łańcucha DNA (tzw. sekwencjonowaniu DNA) jakiego w naturalnej wielkości nie da się przeprowadzić bez pomocy efektywnego algorytmu i odpowiednich programów komputerowych.
W szerszym ujęciu problem przedstawiony w zadaniu polega na odczytaniu łańcucha o długości około 3 miliardów nukleotydów tworzonych przez jedną z czterech zasad: adeninę („A”), guaninę („G”), cytozynę („C”) i tyminę („T”).
Struktura chemiczna DNA
CC BY-SA 3.0
Metoda zarysowana w zadaniu to jeden z komputerowo wspomaganych sposobów zwany „sekwencjonowaniem przez hybrydyzację”. Szkic nie uwzględnia na przykład błędów jakie mogą powstać w trakcie odczytywania sekwencji polegających na pojawianiu się odczytów, którym nie odpowiadają żadne rzeczywiste sekwencje (tzw. błędy dodatnie) lub nieodczytywaniu sekwencji, które powinny być w roztworze (tzw. błędy ujemne). Sekwencjonowanie DNA ma dziś ogromne znaczenie w kryminalistyce, sądownictwie, rolnictwie, archeologii, farmaceutyce i medycynie.
| Domniemane rozwiązanie zadania
Pozostaje uzasadnić, że nie ma krótszego łańcucha. |

Sekwencja DNA stanowi w pewnym sensie program działania mechanizmów w żywych komórkach, w szczególności pewne jej rejony określają jakie białko zostanie wytworzone a inne, w uproszczeniu, kiedy i gdzie (proces regulacji genów). Białka kodowane są za pomocą kodu genetycznego, w którym trójki zasad kodują pojedynczy aminokwas. Pojedyncze białko jest sekwencją aminokwasów, których standardowo wyróżniamy 20. Większość aminokwasów może być zakodowana w DNA na kilka sposobów (kilka różnych trójek zasad koduje ten sam aminokwas).
Sekwencje aminokwasów to inaczej łańcuchy polipeptydowe i mogą one przyjmować złożone struktury przestrzenne. Ustalenie biologicznie aktywnych struktur przestrzennych białek odpowiadających danej sekwencji aminokwasowej jest jednym z ciekawszych problemów z jakim boryka się biologia molekularna wspierana przez tzw. „bioinformatykę strukturalną”. Tutaj z pomocą przychodzą zaawansowane metody informatyczne tzw. uczenia maszynowego. Otóż znane z doświadczeń biologicznych pary: sekwencja aminokwasów i struktura białkowa są podawane na wejście programu uczącego się, który po pełnym cyklu uczenia powinien umieć prawidłowo wskazać strukturę dla zadanej sekwencji lub sekwencję dla zadanej struktury (np. przy projektowaniu leków). W chwili obecnej istnieją ogromne bazy danych gromadzące te odpowiedniki i używane w maszynowym uczeniu. Pomimo tego ciągle jeszcze nam daleko do perfekcyjnego prognozowania struktur białek.
***
Kierunek studiów bioinformatyka jest prowadzony m.in. na Wydziale Informatyki i Telekomunikacji Politechniki Poznańskiej. Na terenie Politechniki działa Europejskie Centrum Bioinformatyki i Genomiki (ECBiG) jako unikalna na terenie Wielkopolski jednostka badawczo-rozwojowa, powstała na bazie konsorcjum zawiązanego pomiędzy Politechniką Poznańską a Instytutem Chemii Bioorganicznej Polskiej Akademii Nauk w Poznaniu.
Informacje o autorze
Andrzej P. Urbański, Instytut Informatyki, Politechnika Poznańska.
Wykłada programowanie gier komputerowych, a w wolnym czasie pisze opowiadania często przekazujące wiedzę bądź motywacje.
Pisze powieść o programowaniu w języku Python.
Autor dziękuje panu Maciejowi Miłostanowi za konsultację treści.
Przypisy
1 J. Błażewicz, Bioinformatyka i jej perspektywy, wykład inauguracyjny, Politechnika Poznańska, 2011. http://www2.andrzeju.pl/wp-content/uploads/2011/11/wyklad_inauguracyjny_2011.pdf
2 J. D. Watson, Podwójna helisa. Historia odkrycia struktury DNA, Warszawa,1995.


{kind=link}
Dustin Wostmann
Eulean Loutit